Objectives
Upon completion of this module topic, you should:
- Understand the growth pattern for the cells you are working with and be able to use microscopy techniques to observe cell growth and morphology.
- Observe cell cultures regularly and keep record of cell growth and morphology.
- Be able to prepare cell feeding media and understand the role that the major reagents in the media play in supporting your cells.
- Be able to aspirate old feeding media from cell cultures, wash cells and feed cells with fresh media.
- Be able to measure the growth and viability of your cells using an inverted phase contrast microscope, the dye trypan blue to detect cell viability, and a hemacytometer chamber to count cells.
- Be able to subculture adherent cells using dissociation agents (trypsin) when they become semi-confluent (also referred to as passaging, harvesting, and splitting cells).
- Be able to appropriately thaw frozen cells and use specialized freezing media and cryopreservation vials to freeze cells.
- Be able to screen cells for contamination.
Part a
Maintenance
This is Part A, Maintenance, under the module topic, Cell Culture Techniques. This topic part has one section: Content Tutorials
Content Tutorials
I. MAINTENANCE – Cell Culture Experimental Overview
Cultures should be examined daily, observing the morphology, the color of the medium and the density of the cells. A tissue culture log should be maintained that is separate from your regular laboratory notebook. The log should contain: the name of the cell line, the medium components and any alterations to the standard medium, the dates on which the cells were split and/or fed, a calculation of the doubling time of the culture (this should be done at least once during the semester), and any observations relative to the morphology, etc.
A. Growth Pattern
Cells will initially go through a quiescent or lag phase that depends on the cell type, the seeding density, the media components, and previous handling. The cells will then go into exponential growth where they have the highest metabolic activity. The cells will then enter into stationary phase where the number of cells is constant, this is characteristic of a confluent population (where all growth surfaces are covered).
B. Subculturing & Harvesting
Cells are harvested when the cells have reached a population density which suppresses growth. Ideally, cells are harvested when they are in a semi- confluent state and are still in log phase. Cells that are not passaged and are allowed to grow to a confluent state can sometimes lag for a long period of time and some may never recover. It is also essential to keep your cells as happy as possible to maximize the efficiency of transformation. Most cells are passaged (or at least fed) three times a week.
1. Suspension culture. Suspension cultures are fed by dilution into fresh medium.
2. Adherent cultures. Adherent cultures that do not need to be divided can simply be fed by removing the old medium and replacing it with fresh medium. When the cells become semi-confluent, several methods are used to remove the cells from the growing surface so that they can be diluted:
Mechanical – A rubber spatula can be used to physically remove the cells from the growth surface. This method is quick and easy but is also disruptive to the cells and may result in significant cell death. This method is best when harvesting many different samples of cells for preparing extracts, i.e., when viability is not important.
Proteolytic enzymes – Trypsin, collagenase, or pronase, usually in combination with EDTA, causes cells to detach from the growth surface. This method is fast and reliable but can damage the cell surface by digesting exposed cell surface proteins. The proteolysis reaction can be quickly terminated by the addition of complete medium containing serum
EDTA – EDTA alone can also be used to detach cells and seems to be gentler on the cells than trypsin. The standard procedure for detaching adherent cells is as follows:
a. Visually inspect daily. b. Release cells from monolayer surface: -Wash once with a buffer solution – Treat with dissociating agent. – Observe cells under the microscope and incubate until cells become rounded and loosen when flask is gently tapped with the side of the hand. – Transfer cells to a culture tube and dilute with medium containing serum. – Spin down cells, remove supernatant and replace with fresh medium. c. Count the cells in a hemacytometer, and dilute as appropriate into fresh medium.
C. Media and growth requirements
1. Physiological parameters – temperature – 37C for cells from homeotherms – pH – 7.2-7.5 and osmolality of medium must be maintained – humidity is required – gas phase – bicarbonate concentration and CO2 tension in equilibrium – visible light, can have an adverse effect on cells; light induced production of toxic compounds can occur in some media; cells should be cultured in the dark and exposed to room light as little as possible
2. Medium requirements: (often empirical) – Bulk ions – Na, K, Ca, Mg, Cl, P, Bicarb or CO2 – Trace elements – iron, zinc, selenium – sugars – glucose is the most common – amino acids – 13 essential – vitamins – B, etc. – choline, inositol – serum, contains a large number of growth promoting activities such as buffering toxic nutrients by binding them, neutralizes trypsin and other proteases, has undefined effects on the interaction between cells and substrate, and contains peptide hormones or hormone-like growth factors that promote healthy growth. – antibiotics, although not required for cell growth, antibiotics are often used to control the growth of bacterial and fungal contaminants.
3. Feeding – 2-3 times/week.
4. Measurement of growth and viability. The viability of cells can be observed visually using an inverted phase contrast microscope. Live cells are phase bright; suspension cells are typically rounded and somewhat symmetrical; adherent cells will form projections when they attach to the growth surface. Viability can also be assessed using the vital dye, trypan blue, which is excluded by live cells but accumulates in dead cells. Cell numbers are determined using a hemacytometer.
5. Observing cells. Everything that the cell plates or flasks come into contact with must be wiped with 70% ethanol. In complying with this, closely follow each step:
- Wipe incubator door and microscope stage with 70% ethanol.
- Wipe gloved hands with 70% ethanol.
- Check cells under microscope.
- Place cells back into incubator.
6. Feeding Cells
- Do steps 1-4. Listed above.
- Wipe laminar hood with 70% ethanol.
- Take out media from refrigerator, wipe down with 70% ethanol and place in the hood.
- Measure out the desired amount of media and pipette into a centrifuge tube.
- Set the centrifuge tube on bench to warm up for at least 15 minutes.
- Put hood UV light for at least 15 minutes.
- Take cells out of the incubator and place inside the hood.
- Wipe media tube with 70% ethanol and place inside the hood.
- Aspirate off existing media from the flask or microplate.
- Pipette fresh media into the flask.
- Check cells under microscope.
- Place cells back in the incubator.
- Wipe surface of hood with 70% ethanol.
- Clean aspirator hose with autoclaved SigmaClean water bath solution.
- Leave UV light on for at least 5 minutes.
7. Subculturing UMR-106 cells (can be modified for other cell lines)
- Take out 0.25% Trypsin-EDTA from -80°C freezer and let it thaw.
- Do steps 1-4 in “Observing Cells” and steps 2-9 in “Feeding Cells”.
- Pipette 6 ml of 0.25% Trypsin-EDTA into flask and incubate for two minutes.
- Check flask under microscope to see evidence of cell detachment.
- Tap bottom of flask lightly to detach more cells.
- Pipette 6 ml of media into flask and pipette up and down against cell wall until cells have become suspended.
- Pipette cell suspension into a 15 mL centrifuge tube.
- Place tube into ultra centrifuge for 5 minutes at 600 rpm with a counter balance.
- Wipe centrifuge tube with 70% ethanol and place back into the hood.
- Aspirate the media, leaving a small layer of media on top of the cell pellet.
- Slowly pipette 5 ml of media into the tube and re-suspend the cells.
- Pipette out 100 ul of media into eppendorf tube.
- Use this eppindorf for cell counting. Pipette out 20 ul of cell suspension and 80 ul of trypan blue into another eppendorf tube. Pipette enough to coat the surface of the hemocytometer.
- Count cells and calculate the number of cells to seed into the flask. (Approximately 1,000,000 cells for a T-75 flask, 20,000 cells/well for a 24 well plate, 3200 cells/well for a 96 well plate.)
- Re-suspend cells and pipette cell suspension into a flask. Add fresh media.
- Check cells under microscope and place in incubator.
- Wipe surface of hood with 70% ethanol.
- Clean aspirator hose with autoclaved SigmaClean” water bath solution.
- Turn on UV light for at least five minutes.
Part b
Tissue Culture Methods
This is Part b, Tissue Culture Methods, under the module topic, Cell Culture Techniques. This topic part has two sections: Content Tutorial & Animation.
Content Tutorials
I. TISSUE CULTURE METHODS
Cells should be monitored daily for morphology and growth characteristics, fed every 2 to 3 days, and subcultured when necessary. A minimum of two, 25 cm2 flasks should be carried for each cell line; these cells should be expanded as necessary for the transfection experiments. Each time the cells are subcultured, a viable cell count should be done, the subculture dilutions should be noted, and, after several passages, a doubling time determined. As soon as you have enough cells, several vials should be frozen away and stored in liquid N2. One vial from each freeze down should be thawed 1-2 weeks after freezing to check for viability. These frozen stocks will prove to be vital if any of your cultures become contaminated. Procedures:
A. Media preparation
When working in a tissue culture facility, you will be responsible for maintaining your own stock of cell culture media; the particular type of media, the sera type and concentration, and other supplements will depend on the cell line. Do not share media with anyone else because if a culture or a bottle of media gets contaminated, you have no back-up. Most of the media components will be purchased prepared and sterile. In general, all you need to do is aseptically combine several sterile solutions. To test for sterility after adding all components, pipette several milliliters from each media bottle into a small sterile petri dish or culture tube and incubate at 37°C for several days. Use only media that has been sterility tested. For this reason, you must anticipate your culture needs in advance so you can prepare the reagents necessary. But, please try not to waste media. Anticipate your needs but don’t make more than you need. Tissue culture reagents are very expensive; for example, bovine fetal calf serum cost ~ $200/500 ml. Some cell culture additives will be provided in a powdered form. These should be reconstituted to the appropriate concentration with double-distilled water (or medium, as appropriate) and filtered (in a sterile hood) through a 0-22 um filter.
All media preparation and other cell culture work must be performed in a laminar flow hood. Before beginning your work, turn on blower for several minutes, wipe down all surfaces with 70% ethanol, and use ethanol wash to clean your hands. Use only sterile pipettes, disposable test tubes and autoclaved pipette tips for cell culture. All culture vessels, test tubes, pipette tip boxes, stocks of sterile eppendorfs, etc. should be opened only in the laminar flow hood. If something is opened elsewhere in the lab by accident, you can probably assume it is contaminated. If something does become contaminated, immediately discard the contaminated materials into the biohazard container and notify the instructor.
B. Growth and morphology
Visually inspect cells frequently. Cell culture is sometimes more an art than a science. Get to know what makes your cells happy. Frequent feeding is important for maintaining the pH balance of the medium and for eliminating waste products. Cells do not typically like to be too confluent so they should be subcultured when they are in a semi-confluent state. In general, mammalian cells should be handled gently. They should not be vortexed, vigorously pipetted or centrifuged at greater than 1500 g.
C. Cell feeding
Use prewarmed media and have cells out of the incubator for as little time as possible. Use 10-15 ml for T-25’s, 25-35 ml for T-75’s and 50-60 ml for T-150’s.
a. Suspension cultures. Feeding and subculturing suspension cultures are done simultaneously. About every 2-3 days, dilute the cells into fresh media. The dilution you use will depend on the density of the cells and how quickly they divide, which only you can determine. Typically 1:4 to 1:20 dilutions are appropriate for most cell lines.
b. Adherent cells. About every 2-3 days, pour off old media from culture flasks and replace with fresh media. Subculture cells as described below before confluency is reached.
D. Subculturing adherent cells
When adherent cells become semi-confluent, subculture using 2 mM EDTA or trypsin/EDTA. Trypsin-EDTA :
- Remove medium from culture dish and wash cells in a balanced salt solution without Ca++ or Mg++. Remove the wash solution.
- Add enough trypsin-EDTA solution to cover the bottom of the culture vessel and then pour off the excess.
- Place culture in the 37ºC incubator for 2 minutes.
- Monitor cells under microscope. Cells are beginning to detach when they appear rounded.
- As soon as cells are in suspension, immediately add culture medium containing serum. Wash cells once with serum containing medium and dilute as appropriate (generally 4-20 fold).
EDTA alone:
- Prepare a 2 mM EDTA solution in a balanced salt solution (i.e., PBS without Ca++ or Mg++).
- Remove medium from culture vessel by aspiration and wash the monolayer to remove all traces of serum. Remove salt solution by aspiration.
- Dispense enough EDTA solution into culture vessels to completely cover the monolayer of cells.
- The coated cells are allowed to incubate until cells detach from the surface. Progress can be checked by examination with an inverted microscope. Cells can be gently nudged by banging the side of the flask against the palm of the hand.
- Dilute cells with fresh medium and transfer to a sterile centrifuge tube.
- Spin cells down, remove supernatant, and resuspend in culture medium (or freezing medium if cells are to be frozen). Dilute as appropriate into culture flasks.
E. Thawing frozen cells
- Remove cells from frozen storage and quickly thaw in a 37°C waterbath by gently agitating vial.
- As soon as the ice crystals melt, pipette gently into a culture flask containing prewarmed growth medium.
- Log out cells in the “Liquid Nitrogen Freezer Log” Book, if applicable.
F. Freezing cells
- Harvest cells as usual and wash once with complete medium.
- Resuspend cells in complete medium and determine cell count/viability.
- Centrifuge and resuspend in ice-cold freezing medium: 90% calf serum/10% DMSO, at 106 – 107 cells/ml. Keep cells on ice.
- Transfer 1 ml aliquots to freezer vials on ice.
- Place in a Mr. Frosty container that is at room temperature and that has sufficient isopropanol.
- Place the Mr. Frosty in the -70ºC freezer overnight. Note: Cells should be exposed to freezing medium for as little time as possible prior to freezing.
- Next day, transfer to liquid nitrogen (DON’T FORGET) and log in the “Liquid Nitrogen Freezer Log” Book, if applicable.
- Perform viable cell counts.
G. Counting Cells – Use of a Hemacytometer
University of Calgary – Reference, P.J. Hansen Dept of Animal Sciences, University of Florida A hemacytometer (also spelled hemocytometer) is an etched glass chamber with raised sides that will hold a quartz coverslip exactly 0.1 mm above the chamber floor. The counting chamber is etched in a total surface area of 9 mm2 (see Figure 1).

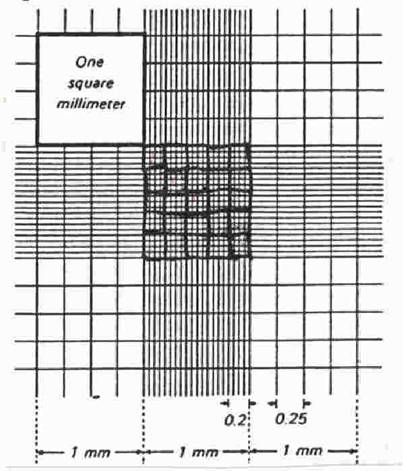
Figure 1: Dimensions of a hemacytometer.
Calculation of concentration is based on the volume underneath the cover slip. One large square (see “W” in Figure 2) has a volume of 0.0001 ml (length x width x height; i.e., 0.1 cm x 0.1 cm x 0.01 cm). In both methods, the hemacytometer is filled by capillary action – place the pipette that is filled with a well-suspended mix of cells at the notch at the edge of the hemacytometer and then slowly expel some contents so that the fluid is drawn into the chamber by capillary action. Staining of cells often facilitates visualization and counting. Either mix cells with an equal volume of trypan blue [0.4% (w/v) trypan blue in PBS] to determine live/dead count (dead cells are blue) or kill cells with 10% formalin and then stain with trypan blue or other another stain (to improve visualization of all cells). Here are two simple methods for counting cells based on the surface area of the hemacytometer used to determine cell count. Other counting schemes are acceptable also. The choice of methods depends upon the cell concentration and the accuracy of the procedure depends upon the number of cells counted. When cell concentration is low, one should count more grids. Method A Count the number of cells in the 4 outer squares (see the left panel of Figure 2). The cell concentration is calculated as follows: Cell concentration per milliliter = Total cell count in 4 squares x 2500 x dilution factor Example: If one counted 450 cells after diluting an aliquot of the cell suspension 1:10, the original cell concentration = 450 x 2500 x 10 = 11,250,000/ml Method B Estimate cell concentration by counting 5 squares in the large middle square (see the right panel in Figure 2). The cell concentration is calculated as follows: Cell concentration per milliliter = Total cell count in 5 squares x 50,000 x dilution factor Example: If one counted 45 cells after diluting an aliquot of the cell suspension 1:10, the original cell concentration = 45 x 50,000 x 10 = 22,500,000/ml
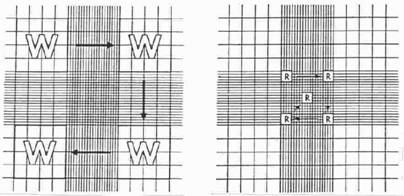
Figure 2. Counting procedure for Methods A (left panel) and B (right panel).
SLIDE PRESENTATION: Cell Counting Baylor College of Medicine: BioEd Online – Lecture Presentation: Measuring and Counting with a Light Microscope, David R. Caprette, PhD. You may view all 14 instructional slides and speaker notes of the presentation, however the focus for cell counting procedures is on the speaker notes and slides 11-14. Slide 11: Counting Chamber (Hemacytometer) Slide 12: Using a Hemacytometer Slide 13: Example: Counting a Cell Suspension Slide 14: Example: Calculating a Cell Concentration
- Cell Counting Slide Presentation (www.bioedonline.org, Multimedia Page)
Animation
Promega Cell-Based Assays – Culture Preparation and Plating for Cell-Based Assays The following video is a narrated experiment that depicts a scientist working in a cell culture room and models how to prepare and plate a cell culture for use in a cell-based assay. Specific techniques that are shown include aseptic technique, washing and feeding cells, subculturing cells, counting cells using a hemacytometer and using centrifugation to harvest cells.
- Culture Preparation Video (www.promega.com, Multimedia Page)