Objectives
Upon completion of this module topic, you will:
- Be able to set up a Polymerase Chain Reaction (PCR) using DNA samples, primers, buffer, dNTPs, and polymerase enzyme. You should be able to explain the significance and role of each component in the reaction.
- Be able to load PCR samples into a thermocycler machine and describe the mechanisms behind the different steps taking place inside of the thermocycler throughout the PCR reaction.
- Be able to explain Real-Time PCR and the two common methods for detection.
- Be able to explain the mechanism of DNA sequencing and identify the advantages of using the Dye Termination method.
Part a
Polymerase Chain Reaction
This is Part A, Polymerase Chain Reaction, under the module topic, Nucleic Acid Amplification & Sequencing. This topic part has three sections: Content Tutorial, Animations, and Activities.
Content Tutorial
DNA Amplification by Polymerase Chain Reaction
The polymerase chain reaction (PCR) is a method of generating many copies of a specific DNA sequence. Newer technologies and sample preparation applications permit the use of PCR in a more timely manner than some conventional technologies. The amount of target nucleic acid in the sample may be extremely small, the amount of the DNA sample is limited such as in forensics and prenatal testing, or when the quality of the DNA sample is poor, yet PCR may yield results not obtainable from other techniques. The procedure will amplify picograms of nucleic acid to micrograms in a short period of time. Because of the specificity and rapidity of the testing, PCR has just about replaced every other NA procedure in clinical labs.
The premise of PCR is based on the DNA polymerase (DNA Pol) enzyme. DNA Pol is the enzyme that incorporates nucleotides onto the end of the replicating DNA strand. Thus DNA Pol needs a DNA template, all four deoxy-nucleotide triphosphates (dNTPs), and free 3’OH groups or primers onto which it adds the new nucleotide in sequence on the base-pairing of dNTP with the template. DNA sequencing does the same thing, but only uses one primer and replicates one strand versus PCR that uses two primers flanking the target sequence and replicates both strands at the same time.
- First Step – DENATURE: The target DNA is denatured (melted) by heating to ≥95°C. The strands remain melted until the temperature is lowered and then base-pairing will reoccur.
- Second Step – ANNEAL: A pair of primers is added in excess quantity to out compete the complementary strand for binding to their target region. The primers are synthetic oligonucleotides constructed to anneal to known sequences flanking either side of the target region. The temperature range for annealing can vary greatly from 40°C to 70°C.
- Third Step – EXTEND: DNA Pol is added to the reaction. It mimics natural replication of DNA and copies an adjacent DNA template to extend across the area between the primers. Extension temperature is commonly 72°C and is based on the type of DNA Pol used.
CYCLE: Heat is again applied to denature DNA, followed by cooling to allow more primer to anneal and primer extension to take place. The reaction takes place in a thermocycler programmed to give rapid and accurate temperature changes. The number of cycles may vary, but most commonly 30-35 cycles are performed. For every one template, 235 copies are theoretically possible.

Don’t panic about all those reagents – all the ingredients are put into the reaction tube prior to the first melt, thus primers and DNA Pol are there for each cycle. The DNA Pol used in most PCR reactions has been isolated from the organism Thermus aquaticus. Taq polymerase is heat stable and able to withstand the repeated heating to 95°C and cooling and still function. Other polymerases are available with other characteristics.
The best graphical representation of PCR is actually animated and found at the DNA Learning Centre (www.dnalc.org, Multimedia Page). You can click on the link, you will need the Shockwave plugin to view it. There will also be several additional animations to view at the end of this PCR tutorial
Amplified DNA (amplicons) may be identified by gel electrophoresis with or without hybridization reactions with probes or used for other investigative procedures. Detection will be described below. The prevention of the amplicons from a completed reaction contaminating new samples is prevented by 1) separate rooms and a one-way work flow and 2) incorporation of dUTP in the dNTP mix rather than dTTP and a special enzyme that degrades any DNA that has dUTP in it. Thus all amplicons that may have contaminated a sample are destroyed at a lower temperature incubation prior to the PCR melt cycle.
Many, many twists on PCR exist. For example, Nested PCR uses a second set of primers nested inside the sites of the first ones to increase specificity of the target region. Semi-nested uses one nested primer and one of the primers from the first reaction. Nested reactions can be done in one step in some instances, but commonly are done in two separate reactions. Multiplex PCR is the amplification of two or more targets in the same reaction tube. In multiplex PCR, the different primer sets need to have the same reaction temperature patterns to get amplification of all the templates. Amplification efficiencies of the different primer/template combinations may require increasing or decreasing the proportion of some primer sets to achieve a balance. Touch-down and touch-up PCR use varying temperatures over the total cycles during the annealing steps. Long-distance PCR is used for products that are much greater than 3-4000 bp in length.
Reverse Transcriptase (RT) PCR
- RT-PCR Animation (www.bio.davidson.edu, Multimedia Page)
In the case of RNA virus identification or detection of the mRNA expression of a certain gene, an additional pre PCR step is performed. Simply, the retroviral enzyme, reverse transcriptase (RT), is used to reverse transcribe the RNA in to cDNA. cDNA is stable for long term storage. Among other things, it is used for expression studies to determine the differences between normal and diseased tissues, or genes relevant during development. You can click on the RT-PCR link above to view an interactive animation of the RT-PCR Methodology.
RT-PCR can be done in one or two reactions. In two step reactions, the RNA is reverse transcribed into cDNA, reaction reagents removed, then the cDNA is used in a PCR reaction. In the one step reactions, everything is added in one tube and special enzymes or enzyme mixes that act as RT and DNA Pol are used and the reaction performed.
Real-Time (rt) PCR and/or Quantitative PCR (qPCR)
Real-time PCR is the fastest growing PCR technology on the market today. The advantages are distinct for both research and clinical applications. rtPCR thermocylers are able to detect the accumulation of DNA amplicons during the cycles by the use of fluorescent dye, SYBR Green, incorporated in the reaction mix. In addition, specific targets can be detected by the use of multiple detection oligonucleotides that also bind to their target DNA. For these types of reactions, increasing fluorescence indicates a positive PCR reaction. Real-time becomes quantitative by using standard template preparations of known numbers of template copies. Standard curves derived from the data of the standard template preps can be used to determine exactly the number of templates in the original sample.



Figures: Standard curves from rtPCR. Each reaction was run in duplicate. RFU = Relative Fluorescent Units.
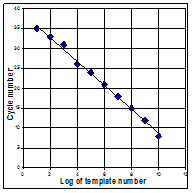
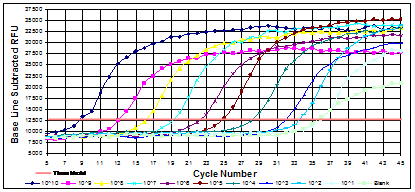
Standards of a high known template numbers will produce product detectable in earlier cycles than those of lower template numbers. A standard curve plots cycle number versus template number. Another advantage to rtPCR is its ability to determine melting temperatures of DNA. CG pairs have 3 bonds holding the strands together whereas TA pairs have two. DNA that has more C+G than A+T will need more heat to melt the two strands in to single ones. When combined with fluorescently-labeled oligoprobes, the latest applications for rtPCR include detection of single nucleotide mutations in genes, identification of specific targets in a mixed population and of interest in microbiology, the ability to identify bacteria based on their melting profiles.
Quantitating Gene Expression: qPCR & Probes
A nucleic acid probe is a short, single-stranded nucleic acid that can be either RNA or DNA and is complementary to part of the nucleotide sequence of a particular nucleic acid region or gene of interest. If part of the nucleotide sequence of the particular gene of interest is known, a probe can be synthesized that is complementary to the region. Probes will hydrogen-bond specifically to a complementary strand in the desired gene and the probe is labeled with a radioactive isotope or a fluorescent tag so that it can be tracked. Probes are valuable tools for quantitating gene expression through real-time qPCR.
With the ability to quantitate templates, real-time qPCR becomes even more powerful in gene expression work. rt/qPCR can, with the addition of probes to specifically identify your desired target (see below), just about replace Northern blots for RNA expression. There may be some more flexibility in some Northern procedures, but rt/qPCR is much more sensitive and faster. Housekeeping gene standards can be done in separate reactions on the same run (SYBR detection) or can be done in multiplex reactions in the same tube (multiple colored probes).
DNA Probes for rt/qPCR:
DNA probes labeled with a fluorescent, chemiluminescent, colorimetric, or radioactive marker can be used to detect an amplicon. These probes are designed to hybridize to a short sequence within the amplicon. In certain cases (e.g. Real-Time PCR), amplification and detection can occur simultaneously within the same reaction vessel. Fluorescent-based probes have gained the most popularity. One can perform a PCR assay to simultaneously detect different targets (i.e. Multiplex PCR) by using probes with different fluorescent markers. Several different probe detection systems are commercially available, and the list is growing.
FRET Probes:
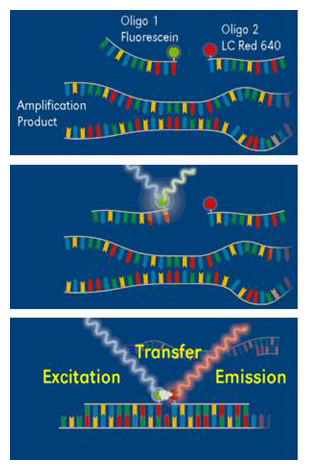 FRET (Fluorescence Resonance Energy Transfer) is a powerful technique for real-time PCR product detection. In addition to the reaction components used for conventional PCR, two specially designed, sequence-specific oligonucleotide probes labeled with fluorescent dyes (FRET probes) are applied for this detection method. This allows highly specific detection of the amplification product. The first dye (fluorescein) is excited by a filtered light source, and emits green fluorescent light at a slightly longer wavelength. When the two dyes are in close proximity, the emitted energy excites the LC Red 640 (or other compatible dye) attached to the second hybridization probe that subsequently emits red fluorescent light at an even longer wavelength. This energy transfer, referred to as FRET, is highly dependent on the spacing between the two dye molecules. Only if the molecules are in close proximity (a distance between 1–5 nucleotides) is the energy transferred at high efficiency. The increasing amount of measured fluorescence is proportional to the increasing amount of DNA generated during the ongoing PCR process.
FRET (Fluorescence Resonance Energy Transfer) is a powerful technique for real-time PCR product detection. In addition to the reaction components used for conventional PCR, two specially designed, sequence-specific oligonucleotide probes labeled with fluorescent dyes (FRET probes) are applied for this detection method. This allows highly specific detection of the amplification product. The first dye (fluorescein) is excited by a filtered light source, and emits green fluorescent light at a slightly longer wavelength. When the two dyes are in close proximity, the emitted energy excites the LC Red 640 (or other compatible dye) attached to the second hybridization probe that subsequently emits red fluorescent light at an even longer wavelength. This energy transfer, referred to as FRET, is highly dependent on the spacing between the two dye molecules. Only if the molecules are in close proximity (a distance between 1–5 nucleotides) is the energy transferred at high efficiency. The increasing amount of measured fluorescence is proportional to the increasing amount of DNA generated during the ongoing PCR process.
These probes are composed of short (about 20-25 bases) oligonucleotides labeled with two different flourescent dyes. There is a reporter dye on the 5′ terminus and a quenching dye on the 3′ terminus. This oligonucleotide probe sequence is homologous to an internal target sequence present in the PCR amplicon. When the probe is intact, energy transfer occurs between the two flourophors and emission from the reporter is quenched by the quencher. During the extension phase of PCR, the probe is cleaved by 5′ nuclease activity of Taq polymerase thereby releasing the reporter from the oligonucleotide-quencher and producing an increase in reporter emission intensity.
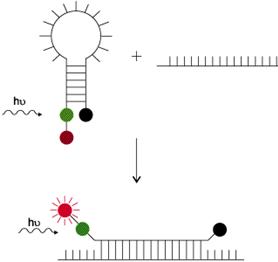
Molecular Beacons:

These are hairpin-shaped oligonucleotide probes with an internally quenched fluorophore whose fluorescence is restored upon binding to a target nucleic acid (diagram below). They are designed in such a way that the loop portion of the molecule is a probe sequence complementary to a target nucleic acid molecule. The stem is formed by the annealing of complementary arm sequences on the ends of the probe sequence. A fluorescent moiety is attached to the end of one arm and a quenching moiety is attached to the end of the other arm. The stem keeps these two moieties in close proximity to each other, causing the fluorescence of the fluorophore to be quenched by energy transfer. Since the quencher moiety is a non-fluorescent chromophore and emits the energy that it receives from the fluorophore as heat, the probe is unable to fluoresce. When the probe encounters the target molecule of interest, it forms a hybrid that is longer and more stable than the stem hybrid and its rigidity and length preclude the simultaneous existence of the stem hybrid. Thus, the molecular beacon undergoes a spontaneous conformational reorganization that forces the stem apart, and causes the fluorophore and the quencher to move away from each other, leading to the restoration of fluorescence which can be detected by a fluorometer (incorporated into real-time PCR instruments).
FRET Beacon or Wavelenght shifting probes . Image: Molecular Probes
Light Upon eXtension – LUX Primers:

LUX Primers are oligonucleotides labeled with a single fluorophore, custom-synthesized according to the DNA/RNA of interest. Typically 20-30 bases in length, they are designed with a fluorophore close to the 3’end in a hairpin structure. This configuration intrinsically renders fluorescence quenching capability; no separate quenching moiety is needed. When the primer becomes incorporated into the double-stranded PCR product, the fluorophore is “dequenched”, resulting in a significant increase in fluorescent signal. This signal increase is the basis for the LUX™ detection platform. [Image and text (modified) from the Invitrogen web site]
Amplifluor UniPrimers:
 In the first round of amplification, the reverse primer, containing a 3′ 18 base oligonucleotide tail (Z sequence) or sequence tag, primes synthesis along the template. In the second round, the forward primer primes synthesis that extends through the special sequence tag, forming a complementary sequence to the tag. In the third round, the UniPrimer hybridizes to the Z sequence (sequence) via the special Z’ sequence (sequence) tag. The hairpin structure of the UniPrimer ensures that the quencher (black circle in figure) suppresses the fluorescence of the fluorophore (green circle in figure). Finally, in the fourth round, synthesis extends through the hairpin loop, relieving the quenching of the fluorophore.
In the first round of amplification, the reverse primer, containing a 3′ 18 base oligonucleotide tail (Z sequence) or sequence tag, primes synthesis along the template. In the second round, the forward primer primes synthesis that extends through the special sequence tag, forming a complementary sequence to the tag. In the third round, the UniPrimer hybridizes to the Z sequence (sequence) via the special Z’ sequence (sequence) tag. The hairpin structure of the UniPrimer ensures that the quencher (black circle in figure) suppresses the fluorescence of the fluorophore (green circle in figure). Finally, in the fourth round, synthesis extends through the hairpin loop, relieving the quenching of the fluorophore.
Scorpion Probes:
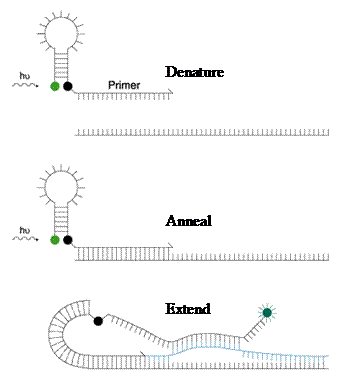 Scorpions are PCR primers with a stem-loop tail, like the molecular beacons, containing a fluorophore and a quencher, but in addition, the primer is attached as well. The stem-loop tail is separated from the PCR primer sequence by a “PCR stopper”. The “PCR stopper” is a chemical modification that prevents the PCR from copying the stem-loop sequence of the Scorpion primer. During PCR, the Scorpion primers are extended to form PCR products. At the appropriate stage in the PCR cycle (the annealing phase), the probe sequence in the Scorpion tail curls back to hybridize to the target sequence in the PCR product. As the tail of the Scorpion and the PCR product are now part of the same strand of DNA, the interaction is intermolecular, making product/probe interactions even faster than that of the molecular beacons.
Scorpions are PCR primers with a stem-loop tail, like the molecular beacons, containing a fluorophore and a quencher, but in addition, the primer is attached as well. The stem-loop tail is separated from the PCR primer sequence by a “PCR stopper”. The “PCR stopper” is a chemical modification that prevents the PCR from copying the stem-loop sequence of the Scorpion primer. During PCR, the Scorpion primers are extended to form PCR products. At the appropriate stage in the PCR cycle (the annealing phase), the probe sequence in the Scorpion tail curls back to hybridize to the target sequence in the PCR product. As the tail of the Scorpion and the PCR product are now part of the same strand of DNA, the interaction is intermolecular, making product/probe interactions even faster than that of the molecular beacons.
The exquisite sensitivity of oligonucleotide primers and probes to mismatched sequences has permitted the development of many assays utilizing the mismatch. Single nucleotide polymorphisms (SNPs) can be detected in genes of known genetic disease, tumor mutation or HLA types. With primers, if the 3’ end of a PCR primer is mismatched, amplification will not occur, making positive/negative a quick PCR protocol used in HLA and antibiotic resistance testing. With probes, the melting characteristics of mismatched probes are not the same as matched probes. rtPCR assays are in use or development using multiple probes and melting characteristics as a way to identify bacteria directly from the specimen. Many of probes described above can be used in rtPCR for detection SNP or mutation detection as well as the more straightforward detection of the amplicons themselves.
One important piece of information about rtPCR is that it is VERY fast. One of the thermocyclers on the market now uses hot air to change the temperature. Runs of 30 cycles can be complete in 1 hour as compared to older thermocyclers that took 3-4 hours. In addition, the detection procedure is built right in, so no other techniques need be employed and post-amplification contamination of new samples is reduced/eliminated as the tubes need not be opened to detect the amplicons. When you add in the ability to do 4 or more colored fluorophores in a multiplex reaction you have PCR that is fast, quantitative, versatile and full of information.
University of Calgary Biotechnology Training Centre
Animations
1. PowerPoint: View the PCR Slides
- PCR PowerPoint (PowerPoint Presentation)
2. Task: Follow the link below to the Gene Almanac of the Cold Spring Harbor Laboratory Dolan DNA Learning Center. The website provides an interactive animation of PCR. The animation provides an introduction to PCR technology and its applications and simulates the PCR mechanism for amplification supplemented with a graphical representation of the amplification process. In addition the animation continues into the technique used for analyzing a PCR product- gel electrophoresis (Module Subset II-a.) along with a further review of gel visualization applications used to detect human DNA polymorphisms (review of RFLP’s).
- Gene Almanac (www.dnalc.org, Multimedia Page)
3. Follow the link below to the DNA Interactive Website. Click on the “Manipulation” section. Next, click on the “Techniques” module and then click on the “Amplifying” section to be taken to the following PCR animations.
- DNA Interactive Website (www.dnai.org, Multimedia Page)
Tasks: a. View the first 2D animation provided: Making many copies of DNA, to learn about the mechanism of the Polymerase Chain Reaction. b. View the second animation which presents a narrated 3D animation of PCR. c. View the interviews to learn about the history and mechanisms behind PCR: Making many DNA copies, Naming PCR, Finding the DNA to copy.
4. PCR Animation 1: Animations from Raven, Johnson, Losos, & Singer, Biology, 7th Edition, 2005 Click on the following link to view the animation list. Now click on the animation titled “Polymerase Chain Reaction”. The animation provides a narration as well as guided text to support the simulation.
- Polymerase Chain Reaction (highered.mcgraw-hill.com, Multimedia Page)
5. cDNA: Complemenatary DNA Animations from Raven, Johnson, Losos, & Singer, Biology, 7th Edition, 2005
Click on the following link to again view the animation list. Now click on the animation titled “cDNA”. This animation will review the processes involved in synthesizing complementary DNA. Be sure to note the role that the enzyme reverse transcriptase plays in the process. This animation will also be useful to reference when you learn about cDNA libraries in module subset II-d.
- Complementary DNA (highered.mcgraw-hill.com, Multimedia Page)
6. PCR Animation 2: This animation provides additional reinforcement of PCR steps in a simplified version.
- PCR Animation #2 (sciencelauncher.com, Multimedia Page
7. Reverse Transcriptase: RT-PCR Animation: This animation depicts the process of RT-PCR emphasizing the role that the enzyme reverse transcriptase plays in the process. You can connect this animation to the role that reverse transcriptase plays in the cDNA animation above.
- Reverse Transcriptase (sciencelauncher.com, Multimedia Page)
Activities
1. PCR Virtual Lab – The University of Utah Genetic Science Learning Center Task: Complete the University of Utah’s Polymerase Chain Reaction Virtual Lab. To access the lab follow the link provided below.
- Polymerase Chain Reaction Virtual Lab (learn.genetics.utah.edu, Multimedia Page)
When you have completed the virtual lab please respond electronically to the following short response question: Describe the essential features of the Polymerase Chain Reaction (PCR) and outline the steps involved in carrying out the application. In addition, explain the overall value of utilizing the PCR technique as an important tool in a molecular biology laboratory.
2. Arizona Biology Project – Recombinant DNA Technology Problem Set In this problem set, you will learn about some of the basic techniques of recombinant DNA, and how recombinant DNA technology is applied to human health. Complete problems 1,6,7,8 from the set. The following problems have multiple choice answers. Correct answers are reinforced with a brief explanation. Incorrect answers are linked to tutorials to help solve the problem.
- Recombinant DNA Technology Problem Set (www.biology.arizona.edu, Multimedia Page)
3. The Polymerase Chain Reaction. Click on the following link to review an animation of PCR and when you are finished identify the 4 main steps involved in the reaction and briefly describe what happens in each step.
- Polymerase Reaction Chain (library.thinkquest.org, Multimedia Page)
Part b
Nucleic Acid Sequencing
This is Part B, Polymerase Chain Reaction, under the module topic, Nucleic Acid Amplification & Sequencing. This topic part has two sections: Content Tutorial, and Activities.
Content Tutorial
DNA Sequencing
PCR, RT-PCR and sequencing are common techniques from the research laboratory to all the clinical laboratory disciplines. As the DNA sequence of a gene/organism IS the sequence, determining the sequence is considered the most accurate protocol you can do. Costs in the past have kept sequencing to the research world and only when required, but automation has increased the throughput, decreased the turn around time and increased the length and quality of the sequence.
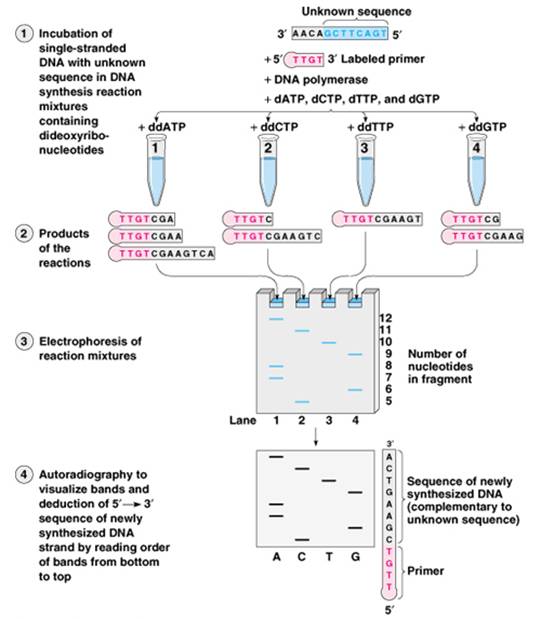
DNA sequencing is the forerunner of PCR and is now the highest resolution way to determine complex gene families such as the HLA (Human Leukocyte Antigen system that contains a large number of genes related to the functioning of the immune system in humans) genes in tissue transplant patients and donors. Databases of sequences of donors can be used to perform virtual crossmatches with unrelated donors. A single primer is used to make copies of one strand of the DNA. Full-length copies though do not enable the determination of the sequence. To accomplish this, special nucleotides are used in the mix. These nucleotides do not have either the 2’ or 3’ OH groups and are called dideoxynucleotides.

The mixture of the 4 for DNA then is ddNTPs. In the sequencing mix, there is a proportion of normal dNTPs and ddNTPs. The ddNTPs are incorporated by chance in place of the dNTP. Without the 3’OH group, no further dNTPs can be incorporated. This method is called chain termination sequencing. To detect the sequence, one of the dNTPs, usually dCTP is radioactive. Four tubes are prepared each with one of the 4 ddNTPs in the dNTP mix. The 4 reactions are then detected by electrophoresis. The sequences that terminated first will be shorter and migrate the furthest. Sequences are read from shortest to longest pieces, going back and forth from reaction to reaction. (See figure below) Again though, technology has surpassed original thought and instead of radioactive nucleotides, 4 different colored fluorophores are used to label the ddNTPs. Thus sequencing is now a single tube reaction much like PCR (cycle sequencing) and automated readers that use gels and laser detectors read the sequence. One HLA specific sequencing system is able to sequence all the HLA genes of one individual in as little as 6 hours. This is now within the realm of practical transplant use.
Sequence based typing implies the comparison to known sequences. Consequently large databases of sequences must be searched to find the matches and thus to type the gene.
Genebank/NCBI (www.ncbi.nlm.nih.gov, HTML Page) is an obvious place, but can lead to confusing results unless the search is refined. Many standalone databases have come under the NCBI umbrella and specialized databases have moved there. For example, OMIM, (www.ncbi.nlm.nih.gov, HTML Page) Online Mendelian Inheritance in Man and SNP; (www.ncbi.nlm.nih.gov, HTML Page) Single Nucleotide Polymorphism. The best choice is to compare your sequence to specialized databases. Examples include: MLST Multi Locus Sequence-based Typing (www.mlst.net, HTML Page), which is a repository for bacterial sequences based on several different genes; IMGT ImMunoGenTics (www.ebi.ac.uk, HTML Page) is an example of an HLA data base, there are also many more.
Figure: DNA Sequencing
Robotics and Automation One final aspect of DNA isolation, preparation, amplification and sequencing set up is robotics. More and more kits are available for the handling of many samples at once using the 96 well footprint and vacuum systems instead of centrifuges. Other systems have automated PCR and detection, and others have sample preparation. In the case that your institution acquires an automated system for some or all of its molecular diagnostics, you should now feel more comfortable with the theory of its operation. Originally, PCR was done in 100 μl reactions. Now reaction volumes can be less than 10 μl. As the sample size decreases, the precision and accuracy of pipetting can only be accomplished by robotics. PCR has gone from 0.5 ml tubes to 0.2 ml tubes. The 0.2 ml tubes fit the footprint of the standard 96 well ELISA (Enzyme-Linked Immunosorbant Assay) or microtitre plate. The whole new science of laboratory robotics is built on this footprint. To increase the workload and the throughput at the same time, laboratory robots may eventually take over the pipetting of the very small volumes suggested.
University of Calgary Biotechnology Training Centre
Activities
Virtual Lab Essential Questions:
What kind of patient samples are used for the purpose of identifying possible pathogens? What does PCR do, how does it work, and why is it useful? How do you separate the desired DNA from all others? How does an automatic DNA sequencer work? Why is it possible to use a DNA sequence to identify bacteria?
Virtual Lab Assignment: Complete the “Bacterial ID Lab” from the Howard Hughes Medical Institute’s Virtual Lab Series. Complete all tasks involved in the lab and respond to the essential questions listed above when you have finished the lab.
- Bacterial ID Lab (www.hhmi.org, Multimedia Page)
Directions: Throughout each exercise, this window will display information explaining what you are doing. All the interactions, however, will be done inside the graphic window to the left. The small white box below the graphic will give you specific instructions on what objects to click on.
Virtual Bacterial ID Lab Credits:
Executive Producer Joseph Perpich, M.D., J.D.
Producer Dennis Liu, Ph.D.
Program Design Satoshi Amagai, Ph.D.
Scientific Advisor David Relman, M.D.
Writers Satoshi Amagai, Ph.D.
Laura Bonetta, Ph.D.
3-D Modeling Bob Buffington
Bill Pietsch
Graphics and Interface Design Bill Pietsch
Animation and Programming Davey Thomas
Special Thanks Timothy Wright, Ph.D.